9 Training our first Protein Language Model
In this chapter, we’ll train a protein language model. To run a full training session, you will need a powerful GPU setup or access to cloud computing services, the script below trains on GPCR proteins only to ensure we can study protein language models on a MacBook or moderately powerfull workstation. A Google Colab notebook to train the model is available here.
If you lack the necessary computing resources, you can download a pre-trained version of GPCR-BERT here
All scripts for this chapter are found here: https://github.com/MichelNivard/Biological-language-models/tree/main/scripts/Proteins/Chapter_3
9.1 Introduction
Now that we have prepared a dataset of protein sequences, we can proceed to train a protein language model using the (Modern)BERT model architecture. Protein sequences, like DNA, follow structured patterns that can be learned by deep learning models. This chapter introduces the training of GPCR-BERT, a transformer-based masked language model (MLM) designed to understand and generate protein sequences.
Because the fundamentals of training a Protein language model aren’t that different from training a DNA language model this chapter is somewhat abbreviated, see Chapter 2 for more context, most of which directly translates to protein language models.
9.2 Tokenisation
Protein sequences are represented using the FASTA format, which encodes amino acids with standard single-letter codes. For this model, we define a vocabulary that includes:
- The 20 standard amino acids (A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V)
- Ambiguity codes: B (N/D), Z (Q/E), X (any)
- A gap character: ‘-’
A custom tokenizer is developed to handle these sequences efficiently. The tokenizer splits sequences using the defined vocabulary and applies a WordLevel tokenization approach for better generalization.
import torch
import wandb
from tokenizers import Tokenizer
from tokenizers.models import WordLevel
from tokenizers.pre_tokenizers import Split
from transformers import PreTrainedTokenizerFast, ModernBertConfig, ModernBertForMaskedLM, Trainer, TrainingArguments, DataCollatorForLanguageModeling
from datasets import load_dataset
# Initialize Weights & Biases
wandb.init(project="bert-protein", name="bert-protein-GPCR-training_v1")
# --------------------------------
# Protein Tokenizer with Full FASTA Amino Acid Code
# --------------------------------
# Define vocabulary to include all FASTA amino acids and special symbols
protein_vocab = {
"A": 0, "R": 1, "N": 2, "D": 3, "C": 4, "Q": 5, "E": 6, "G": 7,
"H": 8, "I": 9, "L": 10, "K": 11, "M": 12, "F": 13, "P": 14, "S": 15,
"T": 16, "W": 17, "Y": 18, "V": 19, # Standard 20 amino acids
"B": 20, "Z": 21, "X": 22, # Ambiguity codes: B (N/D), Z (Q/E), X (any)
"-": 23, # Gap character
"[UNK]": 24, "[PAD]": 25, "[CLS]": 26, "[SEP]": 27, "[MASK]": 28
}
# Create tokenizer
protein_tokenizer = Tokenizer(WordLevel(vocab=protein_vocab, unk_token="[UNK]"))
protein_tokenizer.pre_tokenizer = Split("", "isolated") # Character-level splitting
# Convert to Hugging Face-compatible tokenizer
hf_protein_tokenizer = PreTrainedTokenizerFast(
tokenizer_object=protein_tokenizer,
unk_token="[UNK]",
pad_token="[PAD]",
cls_token="[CLS]",
sep_token="[SEP]",
mask_token="[MASK]"
)
print(hf_protein_tokenizer.tokenize("MEEPQSDPSV")) # Test with a short sequence9.3 Load the training data
Unlike large-scale protein language models trained on massive datasets from UniProt or AlphaFold, our approach focuses on a more specialized subset: G-protein coupled receptor (GPCR) genes, sourced from RefProt 90. This choice is driven by computational constraints and the goal of achieving high-quality representations within a biologically meaningful domain. By narrowing our scope, we ensure that the model learns from a well-defined set of sequences, optimizing for accuracy and relevance in GPCR-related research rather than sheer scale.
you’ll find the dataset here, but be aware this isn’t a curated dataset, if you would truly want to train a GPCR transformer, you’d go and spend months studying the best resources for validated GPCR proteins, like the GPCR database and related experimental literature.
# --------------------------------
# Load and Tokenize the Protein Dataset
# --------------------------------
dataset_name = "MichelNivard/UniRef90-GPCR-Proteins" # Generic example for now, will clean own data later
dataset = load_dataset(dataset_name, split="train")
# Shuffle the dataset
dataset = dataset.shuffle(seed=42)
print("dataset loaded")
column_name = "value"
def tokenize_function(examples):
return hf_protein_tokenizer(examples[column_name], truncation=True, padding="max_length", max_length=512)
# Tokenize dataset
tokenized_dataset = dataset.map(tokenize_function, batched=True, remove_columns=[column_name])
# Save tokenized dataset
tokenized_dataset.save_to_disk("tokenized_protein_dataset")
print("dataset tokenized")9.4 Model Architecture & Training
After tokenizing the dataset, we proceed to train the model,
We use the ModernBertForMaskedLM model from the transformers library. The key components of the training setup are:
- Model Configuration:
ModernBertConfigdefines the model’s parameters, including the number of transformer layers and attention heads. - Data Collation:
DataCollatorForLanguageModelingis used to prepare masked language modeling inputs. - Training Arguments: Configured with learning rate scheduling, batch size, and gradient accumulation settings.
- Trainer: The
Trainerclass orchestrates training, validation, and logging usingwandb(Weights & Biases).
# --------------------------------
# Load and Tokenize the Protein Dataset
# --------------------------------
dataset_name = "MichelNivard/UniRef90-GPCR-Proteins" # Generic example for now, will clean own data later
dataset = load_dataset(dataset_name, split="train")
# Shuffle the dataset
dataset = dataset.shuffle(seed=42)
print("dataset loaded")
column_name = "value"
def tokenize_function(examples):
return hf_protein_tokenizer(examples[column_name], truncation=True, padding="max_length", max_length=512)
# Tokenize dataset
tokenized_dataset = dataset.map(tokenize_function, batched=True, remove_columns=[column_name])
# Save tokenized dataset
tokenized_dataset.save_to_disk("tokenized_protein_dataset")
print("dataset tokenized")
# --------------------------------
# Define the BERT Model from Scratch
# --------------------------------
config = ModernBertConfig(
vocab_size=len(protein_vocab),
hidden_size=512,
num_hidden_layers=24,
num_attention_heads=24,
intermediate_size=1024,
max_position_embeddings=512,
type_vocab_size=1,
)
config.pad_token_id = protein_vocab["[PAD]"]
model = ModernBertForMaskedLM(config)
# --------------------------------
# Training Configuration (Prints Loss)
# --------------------------------
training_args = TrainingArguments(
output_dir="./bert-protein",
overwrite_output_dir=True,
logging_steps=1, # Log loss every step
save_steps=1000,
save_total_limit=2,
per_device_train_batch_size=12,
gradient_accumulation_steps=1,
num_train_epochs=3,
learning_rate=5e-4,
weight_decay=0.01,
push_to_hub=False,
report_to="wandb", # wandb logging
)
# MLM Data Collator (Automatically Creates `labels`)
data_collator = DataCollatorForLanguageModeling(tokenizer=hf_protein_tokenizer, mlm=True, mlm_probability=0.15)
# --------------------------------
# Train the Model
# --------------------------------
trainer = Trainer(
model=model,
args=training_args,
train_dataset=tokenized_dataset,
tokenizer=hf_protein_tokenizer,
data_collator=data_collator,
)
trainer.train()
# Save the final model and tokenizer
trainer.save_model("./bert-protein")
hf_protein_tokenizer.save_pretrained("./bert-protein")
print("🎉 Training complete! Model saved to ./bert-protein")
# Save the final model and tokenizer to Hub
model.push_to_hub(repo_id="MichelNivard/GPCRBert-v0.1",use_auth_token="YOUR-TOKEN-HERE")
hf_protein_tokenizer.push_to_hub(repo_id="MichelNivard/GPCRBert-v0.1",use_auth_token="YOUR-TOKEN-HERE")9.5 Result
The model is able to learn the structure of GPCR proteins, thought eh strong variability in loss over batches suggests its not uniformly learning all members of the cluster, perhaps due to data imbalances (see Figure 1).
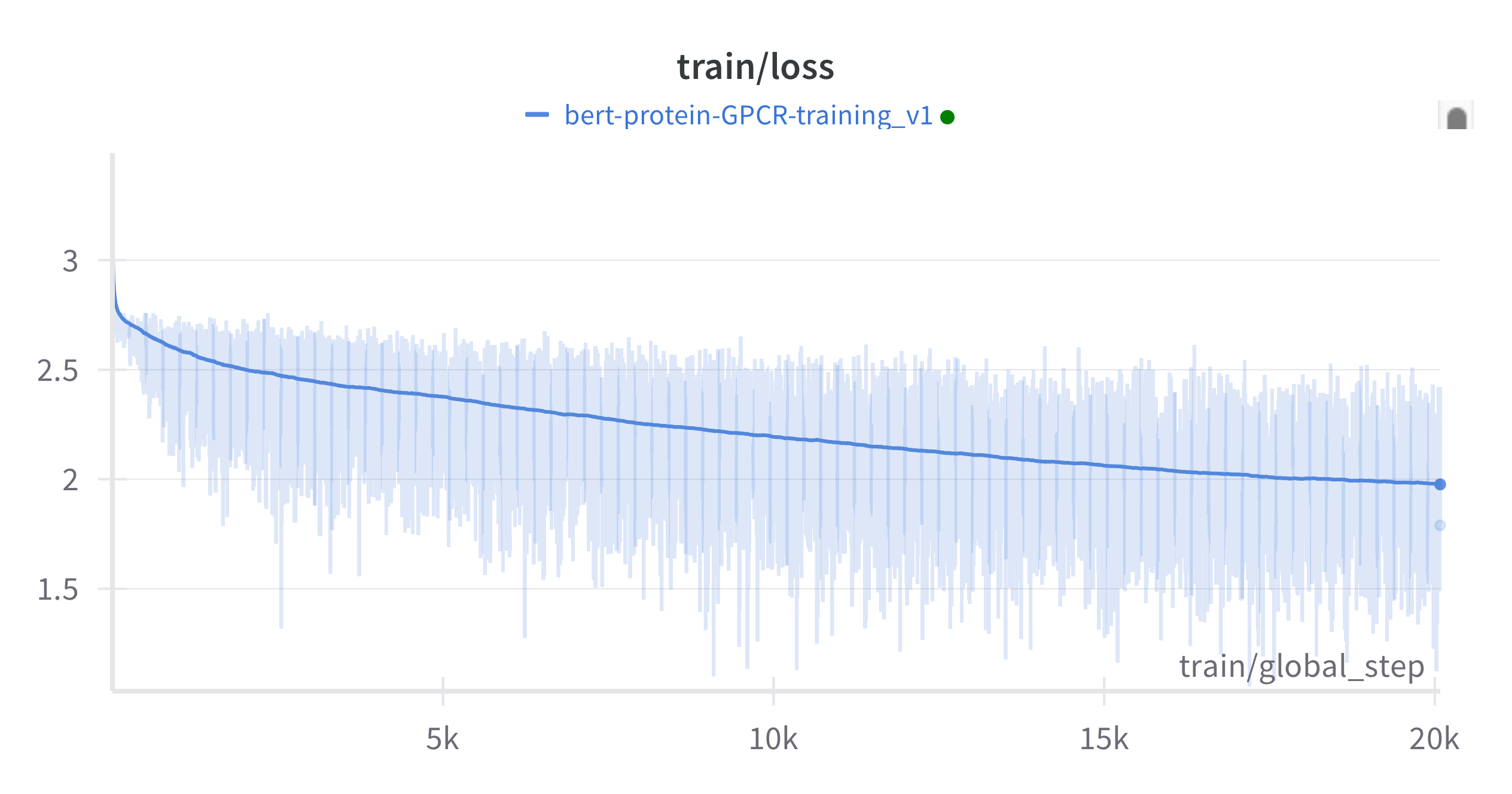
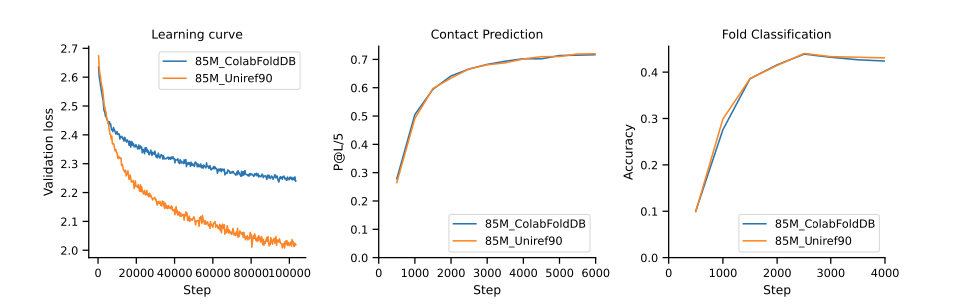
Its interesting to note that the GCPR-Bert training loss is lower then that of much larger protein models trained on larger datasets. This makes sense, the loss (especially on training data!) is highly dependent on the diversity of the data, learning ALL proteins, is harder then learning a subset of proteins. Even at larger scales this phenomena is still evident. In Figure 2 (source (Cheng et al. 2024) Figure A11) we see two love curves for 85m parameter protein language models. Trained on UniRef90 (representative but not extremely diverse) and ColabFoldDB which contains a lot of meta-genomic protein sequences with far greater diversity. Its harder for the model to learn more diverse data, have the higher loss. However, on downstream tasks like atomic contact prediction and fold classification, the models perform nearly identical!
9.6 Homework
Cheng et al. reveal something profound about model loss and scientist’s goalsin their analysis. The loss isn’t a direct measure of the model’s quality for your specific goals. If you are to design a protein language model for a specific tasks (predict the fold of human proteins after mutation for example) then you have to design validation accordingly. As we read in this chapter, and last, that we are running out of animal and plant reference protein data to train protein language models, people have therefore started too include meta-genomic protein data is training. Thets probably a great idea, but its worth considering weather for YOUR downstream tasks it is. ESM3 is better then ESM2, is its trained on way more data, but is it better for your applications?
An interesting unsolved issue to work on in protein model training is how to introduce low diversity but critical protein data in training. We saw models do well on diverse data, but what if we want to introduce data from many individual humans, with disease status as meta info for example, that would be very low diversity data. But the diversity that does exists tells us important things about what variation is, and isn’t tolerated. We need ways to fine-tune a pre-trained models on high value but low diversity data without reducing the models performance. would you reduce the learnign rate, would you mix in very diverse sequences? Would you freeze parts of the model in place?
9.7 Conclusion
This chapter introduced GPCR-BERT, a transformer-based language model for protein sequences. In subsequent chapters, we will explore fine-tuning strategies and downstream applications, such as protein structure prediction. In order to do so we’ll have to really unpack the internals of transformer models, the self-attention mechanism. We’ll return to training protein language models in the chapter “putting it all together” in which we putt all lessons together to do one massive training run and training a protein language model that rivals facebook ESM-2 (and cost me ±900$ out of my own pocket in compute).